
A 58-year-old lady presents with an episode of erythematous papulovesicular rash, which first appeared as weeping blisters on her back after an episode of sunburn when she was 18.
Over time, new lesions have developed on multiple body regions. She reports the condition worsens in summer.
She also reports experiencing dysuria and dyschezia. There is a significant family history, with her sister, nephew and father affected by similar skin lesions.
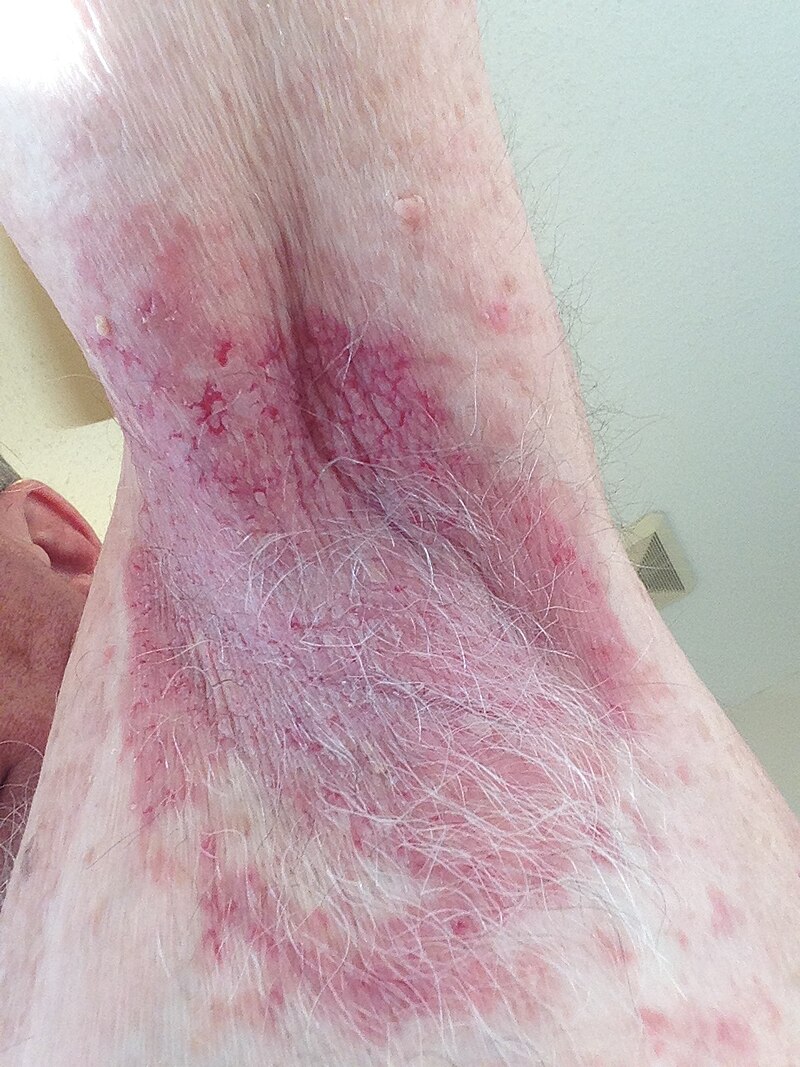
On examination, there are erythematous plaques with scaly crusts, symmetrically distributed on the chest and arms. The groin and inframammary folds are also involved.
These painful and pruritic lesions are clearly demarcated and thickened. Erosions have resulted from the coalescence of lesions. The mucous membranes are spared.
What is the diagnosis?
Correct!
Hailey-Hailey disease (HHD) is an autosomal dominantly inherited disease with complete penetrance but variable expressivity.
The incidence rate is approximately one in 50,000, with males and females equally affected.2 The age of onset is typically during the third or fourth decade.
HHD is caused by ATP2C1 gene mutations located on the long arm of the chromosome,3 which encodes hSPA1C — a calcium ATPase on the Golgi apparatus.
The hSPA1C causes acantholysis by junctional protein sequestration. Adherens junction formation is also impaired due to decreased ATP production, resulting from increased cytoplasmic calcium in keratinocytes.
Moreover, wound healing is impaired in HHD patients as a result of accelerated keratinocyte differentiation due to DNA damage induced by increased reactive oxygen species.
HHD lesions typically occur in intertriginous areas. The inframammary folds and vulva are commonly affected in females.
Characteristic features include symmetrical blisters and erosions that develop on erythematous plaques with pain, pruritus and burning.
The lesions spread outward in an annular pattern. Crusting, painful fissures and malodorous vegetations often develop. The lesions are prone to infections.
Poor healing causes scarring and post-inflammatory hyperpigmentation in affected regions. HHD also causes longitudinal leukonychia.
Diagnosis is by clinical examination and biopsy. Diagnostic histological features include widespread acantholysis, which gives a characteristic ‘dilapidated brick wall’ appearance, lymphohistiocytic infiltration and mild dyskeratoses.
Management involves a combination of lifestyle modifications and medical and/or surgical therapy. Patients are advised to avoid friction, heat and moisture, by avoiding hot environments and wearing loose clothing with absorbent pads.
First-line treatments aim to reduce inflammation and prevent infection. These include topical corticosteroids or calcineurin inhibitors, and topical antimicrobials or antifungals.
More severe disease may require systemic agents, including oral antibiotics, dapsone 2.5-3.5mg/kg/day and methotrexate 15mg/week.
For relapsing cases, oral magnesium supplementation and oral low-dose naltrexone 1.5-4.5mg have shown high efficacy.
Oral immunosuppressive or biologic drugs have been used to achieve sustained remission. Lastly, surgical options like cryotherapy and excision with grafting may be indicated for localised lesions that are not responsive to conservative treatments.